Research News
Watching the Dance of Molecules in the Cloud
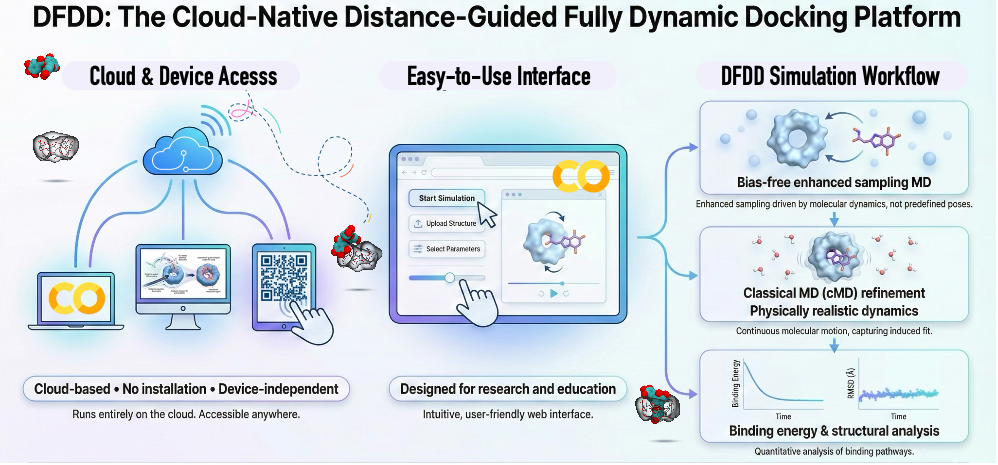
Anyone can do dynamic docking, watching molecules bind in motion.
Researchers at the Center for Computational Sciences, University of Tsukuba, have developed an accessible platform to overcome the limitations of conventional static docking simulations, offering new avenues for education, training, and reproducible research in molecular recognition and supramolecular chemistry. Their Distance-Guided Fully Dynamic Docking (DFDD) platform is a cloud-ready simulation framework that enables students and researchers to explore dynamic docking, visualize molecular binding in motion, and understand how host-guest crystal structures emerge from molecular interactions.
Tsukuba, Japan—Understanding how molecules bind is central to chemistry, biology, and drug design. Nevertheless, most existing tools provide either static snapshots or require computational resources beyond the reach of many researchers and students. Molecular recognition is driven by continuous motion, with flexibility and solvent-mediated interactions playing key roles. However, molecular binding is often interpreted from static crystal structures in conventional docking methods, which typically treat molecules as rigid objects, resulting in many binding pathways remaining poorly captured. While the classical molecular dynamics (cMD) simulations can describe these continuous processes, their high computational cost and limited reproducibility constrain their widespread use.
This work demonstrates how realistic binding pathways and crystal-like structures can be accurately captured using short, reproducible simulations that run on accessible cloud platforms, also in the tablet, offering a practical framework with immediate applicability to learning, teaching, and research. Distance-Guided Fully Dynamic Docking (DFDD) reproducibly recovers binding structures that closely match experimentally determined crystal structures, while also revealing transient configurations that are inaccessible using static docking. DFDD achieved faster convergence, higher reproducibility, and substantially reduced computational effort compared to cMD simulations initiated from unbound states.
DFDD eliminates the need for specialized hardware or complex software installation. It is designed to run on widely accessible cloud platforms, such as Google Colab. By bridging static structural models and fully dynamic simulations, DFDD provides a practical educational and research framework, making dynamic docking accessible to all. With DFDD, everyone can watch molecules bind as crystal-like structures emerge from molecular motion. DFDD is freely available at: https://github.com/nyelidl/DFDD.
###
This research was supported by JSPS KAKENHI Grant Numbers 21H05269 and 24K20888, the JST-CREST project (Grant Number JPMJCR20B3), and the Center for Quantum Information and Life Science (QiLS) at the University of Tsukuba.
Original Paper
- Title of original paper:
- DFDD: A Cloud-Ready Tool for Distance-Guided Fully Dynamic Docking in Host-Guest Complexation
- Journal:
- Journal of Chemical Information and Modeling
- DOI:
- 10.1021/acs.jcim.5c02852
Correspondence
Assistant Professor HENGPHASATPORN Kowit
Assistant Professor Lian Duan
Associate Professor HARADA Ryuhei
Professor SHIGETA Yasuteru
Center for Computational Sciences, University of Tsukuba
Related Link
GitHub: https://github.com/nyelidl/DFDD
Google Colab:
https://colab.research.google.com/drive/1FfTuVSykgsstjzN0nJN0ZQo1_tw0WXSe?usp=sharing
Center for Computational Sciences